
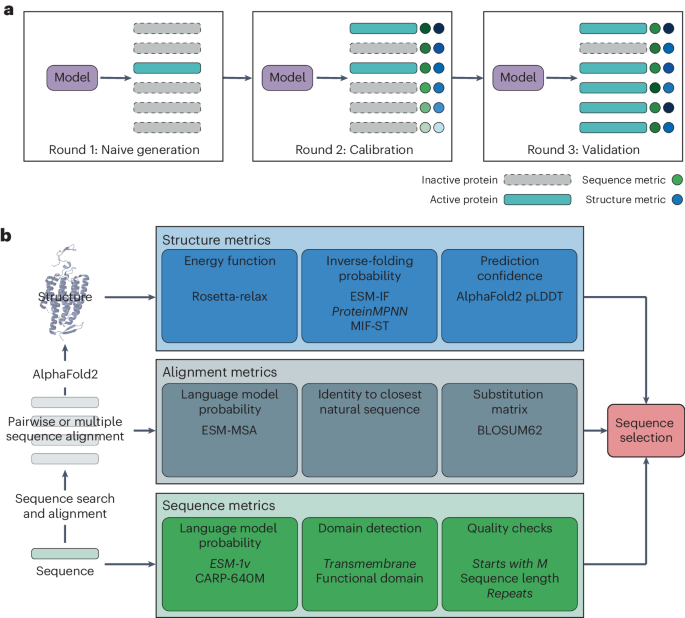
请参阅补充方法以了解全文的其他详细信息。
数据管理
第一轮 CuSOD
尤尼普罗特74 恰好包含一个 Sod_Cu Pfam 的序列75 域已下载。 嗯搜索(嗯, http://hmmer.org/; 参考号 76)确定了 Sod_Cu 域包络。 序列被截断以去除超出 Sod_Cu 匹配范围的无关序列。 进行了额外的质量过滤。 使用 CD-HIT 删除序列重复项77 同一性阈值为 80%,80% 和 20% 分别被随机分为“训练”集和“测试”集。 使用 MUSCLE (v3.8) 通过迭代过程生成训练 MSA78。
第一轮MDH
下载包含 Ldh_1_N Pfam 域和后跟 Ldh_1_C 域的所有 UniProt 序列。 LDH 和 MDH 酶,基于酶委托编号79、LDH 为 1.1.1.27 和 MDH 为 1.1.1.37,从 SwissProt 下载。 MUSCLE 和 hmmbuild 用于构建两组的轮廓隐马尔可夫模型。 Hmmsearch 用于根据 MDH 和 LDH 配置文件对每个 UniProt Ldh_1_N/Ldh_1_C 序列进行评分,并保留与 MDH 配置文件具有更强匹配的序列。 额外的处理与第一轮 CuSOD 数据管理完全相同。
领域架构的量化
请参阅补充方法。
第 2 轮 CuSOD 预测试
UniProt CuSOD 蛋白的获得如上所述(第 1 轮 CuSOD)。 每个序列的起源界均从 UniProt 注释中获得。 使用 Phobius 预测跨膜结构域和信号肽66。 具有跨膜结构域的序列被丢弃。 从预测含有信号肽的序列中去除信号肽。 手动选择一组 14 个代表性 CuSOD 和 2 个 FeSOD 蛋白进行实验筛选,包括预测不含信号肽的真核、病毒和细菌蛋白,以及去除预测信号肽的细菌蛋白。
第 2 轮和第 3 轮 CuSOD
MDH 第 2 轮和第 3 轮
嗯搜索76 用于搜索 Magnify82 对于恰好包含一个 Ldh_1_C 和一个 Ldh_1_N 结构域的序列。 Mgniify 蛋白质列表已添加到 UniProt 列表中(如上所述的管理)。 进行了额外的质量过滤。 使用 CD-HIT 对序列进行 90% 的重复数据删除。 根据 usearch search_global,与第一轮中实验筛选的序列同一性大于 85% 的序列被丢弃。 其余序列分为训练集 (90%) 和测试集 (10%)。 生成了训练 MSA。
系统发育树
使用 FastTree 从 MAFFT 生成的 MSA 构建树83。 树木有根并使用 ETE3 渲染中点(参考文献 1)。 84)。
分支酸变位酶和溶菌酶
请参阅补充方法。
生成模型
ESM-MSA-1b 采样
使用 ESM-MSA-1b 模型通过迭代掩蔽和采样生成序列48。 ESM-MSA-1b 是一种神经网络模型,经过训练可填充蛋白质 MSA 的掩蔽位置中的野生型氨基酸。 该模型可用于通过迭代运行 MSA 掩蔽和预测来生成新序列,每次用从模型返回的概率分布中提取的氨基酸替换掩蔽位置处的野生型氨基酸。 使用掩码语言模型来生成新序列首先由 Wang 和 Cho 提出50,并且该策略已在至少三项先前的工作中应用于蛋白质序列22,49,85。
有关所用参数的更多详细信息,请参阅补充方法。
蛋白质GAN
使用 CuSOD 和 MDH 的训练集训练生成对抗模型。 然后,对于每个家族,通过使用截断正态分布从潜在空间采样向量来生成序列。 在第 1 轮和第 2 轮中,为每个家族生成了 10,048 个序列。 对于第 3 轮,分别为 CuSOD 和 MDH 生成了 560,016 和 160,064 个序列。
祖先序列重建
使用 FastTree 从训练集参考 MSA 生成最大似然树86。 使用 GRASP 的联合重建功能从树中生成祖先序列重建28 命令行工具。 计算指标,并从整个重建序列集中选择候选者。
普罗根
请参阅补充方法。
计算指标
阿尔法折叠2
AlphaFold2(参考。 44)用于预测测试序列和所有通过第一个过滤步骤的生成序列的结构。
福比乌斯
福比乌斯66 (https://phobius.sbc.su.se/data.html) 可执行文件用于预测信号肽或跨膜结构域的存在。
ESM-1v 和 CARP-640M
根据 ESM-1v 计算的分数39 和CARP-640M68 模型是每个位置氨基酸的对数概率的平均值。 在没有掩蔽的情况下,可以通过对每个序列进行一次前向传递来完成此计算。 对于部分掩蔽,可以通过相当于每个 masked_fraction 一次的遍数来完成。
环境管理-MSA
ESM-MSA-1b 的分数48 模型的计算方式与 ESM-1v 分数类似,使用整个序列的平均对数概率。 该指标是使用 phmmer 计算的76 要找到与每个查询最接近的 31 个训练序列,请将 32 个序列与 MAFFT 对齐,并计算掩蔽间隔为 6 的六次传递的平均对数概率。
蛋白质MPNN、ESM-IF 和 MIF-ST
蛋白质MPNN45 和ESM-IF46 分数是使用 AlphaFold2 预测结构的查询残基的平均对数似然。 MIF-ST47 使用蛋白质序列模型存储库中的 extract_mif.py 脚本计算分数(https://github.com/microsoft/ Protein-sequence-models)。
罗塞塔-放松
罗塞塔 (v2020.08.61146)43 松弛程序用于松弛 AlphaFold2 结构。
到最近训练序列的距离
使用 FASTA 包中的 ggsearch36 找到了最相似的训练序列87、BLOSUM62 评分矩阵以及 10 的空位开放罚分和 2 的空位延伸罚分。然后根据查询和顶部命中序列之间的空位比对计算汉明距离。 身份计算为 1 − Hamming_distance。
BLOSUM62 和 PFASUM15 突变体位置平均值
如上所述,使用 ggsearch36 找到了最接近的训练序列。 从比对到最接近的训练序列,平均 BLOSUM62 分数37 计算所有不匹配位置的位置,忽略查询或参考有间隙的位置。 我们还使用替代矩阵 PFASUM15 矩阵计算了比对和分数67。
最长重复次数
计算每个序列中最长的单氨基酸重复和最长的2聚体、3聚体和4聚体重复的分数。 分数计算为-1⨯ 重复单元的数量。 因此,序列 AAAAAA 的单氨基酸重复得分为 -6,2 聚体得分为 -3,3 聚体得分为 -2,4 聚体得分为 -1。 序列 LALALALA 的 1 聚体得分为 -1,2 聚体得分为 -4,3 聚体得分为 -1,4 聚体得分为 -2。
现在
SASA、极性 SASA 和非极性 SASA 是使用 freesasa 软件包根据 AlphaFold2 预测的结构计算得出的(https://freesasa.github.io/)。 使用公式 100 计算极性 SASA 的百分比⨯ 极地现在/现在。
净电荷、Abs(净电荷)和带电分数
通过分别将负电荷和正电荷的谷氨酸和天冬氨酸残基以及赖氨酸和精氨酸残基的数量相加来计算电荷。
平均(phmmer 前 30 名)
phmmer 前 30 名平均分数是通过对训练序列运行实验测试序列的 phmmer 搜索并对前 30 名命中的分数进行平均来计算的。
选择体外测定的序列
第1轮
所选序列与最接近的训练序列具有 70% 和 80% 的同一性,并且 ESM-1v 指标的得分不同。
第2轮预测试
第二轮
所选序列与最接近的训练集序列具有 80% 到 90% 的同一性,并且 ESM-1v 和 ESM-MSA 指标的得分不同。 还通过手动检查对序列进行过滤,以去除那些与最接近的参考序列或长重复序列相比具有较大插入或缺失的序列,并在一些序列的开头添加甲硫氨酸。
第三轮
序列是根据一系列过滤器选择的。 第一个过滤器删除了与最接近的训练序列具有小于 50% 或大于 80% 同一性的序列; (2) 与测试序列相比,ESM-1v 得分低于前 10 个百分位阈值; (3)无起始蛋氨酸; (4)预测的跨膜结构域; (5) 长于三个氨基酸的单氨基酸重复或长于四个氨基酸的氨基酸对重复,因为重复在 ESM-MSA 生成的序列中比在天然序列中更常见(补充图 1)。 35)。 对于每个酶家族,从通过第一级过滤器的序列中随机选择200个ESM-MSA生成的序列和200个GAN生成的序列,并使用AlphaFold2预测它们的结构。 计算每个结构的 ProteinMPNN 分数,并保留每个模型-酶组合中分数最高的 40 个序列。 在前 40 个序列中,随机选择 18 个进行表达和功能表征。 对于选择用于功能表征的每个通过序列,从未通过序列过滤器的序列列表中选择相应的控制序列。 控制序列与最接近的训练序列相同,误差在通过序列的 1% 以内。
新生成的 ProGen 溶菌酶序列
请参阅补充方法。
实验分析
细菌菌株、质粒和生长条件
E. 大肠杆菌 本研究使用BL21(DE3)作为MDH和SOD表达的宿主菌株。 细胞在 37°C 的 LB 培养基上生长,并补充 100 μg ml−1 氨苄青霉素(目录号171254,默克)。
序列优化基于 E. 大肠杆菌-使用 Twist Bioscience 网络界面的首选密码子(www.twistbioscience.com)。 在所有基因的5'末端添加由核糖体结合位点序列和间隔区组成的30bp序列(TTTGTTTAACTTTAAGAAGGAGATATACAT)。 基因从 Twist Bioscience 订购为 pET-21(+) 中 EcoRI 和 NotI 位点之间的克隆。
截短控制序列的质粒构建
请参阅补充方法和补充表 7.
感受态细胞制备和质粒转化
感受态细胞 E. 大肠杆菌 BL21(DE3)采用氯化钙法制备88。
有关详细信息,请参阅补充方法。
蛋白质表达和纯化
通过将过夜培养物以 1:30 的比例稀释到 2.5 ml 自诱导 Terrific Broth (TB) 培养基(包含微量元素(目录号 AIMTB0210,Formedium))中并补充 100 µg ml−1 24 孔格式的氨苄西林。 所有细胞均在 Eppendorf ThermoMixer C 的 24 孔板中培养。为了表达 MDH,细胞在 37°C 下生长 4 小时,然后在 16°C 下生长过夜,同时以 200 rpm 摇动。 为了表达 SOD,细胞在 37°C 下生长 4 小时,然后在 25°C 下以 200 rpm 的转速再培养 3 小时。
3,000 离心收集细胞G 10分钟。 将细胞沉淀悬浮于 200 μl BugBuster 试剂(目录号 70584,Merck)中,并补充有 1 μl 2,000 U ml−1 DNase I(目录号 79254,Qiagen)并在 37°C 下以 200 rpm 摇动孵育 30 分钟。 孵育后,分装 10 μl 混合物并保存在 -20 °C 作为凝胶电泳的总蛋白 (T) 样品。 将混合物以最大速度离心 10 分钟,并丢弃沉淀。 然后,分装10μl上清液并保存在-20℃作为可溶性蛋白(S)样品用于凝胶电泳。 使用以下程序将上清液用于蛋白质纯化。
Talon 树脂(目录号 635653,Takara Bio)用结合缓冲液(50 mM NaH2后4,300 mM NaCl,10 mM 咪唑,pH 7.4),然后悬浮在与树脂床量相同体积的结合缓冲液中。 将 Talon 树脂 (50 µl) 装入 Pierce 微离心柱(目录号 89879,ThermoFisher)中。 将每个上清液样品添加到装载的柱中,并在热混合器中于 4°C 下孵育 30 分钟。
然后将柱在 20 下离心G 30 s,丢弃流动废物。 用 600 µl 洗涤缓冲液洗涤树脂 3 次(50 mM NaH2后4,300 mM NaCl,20 mM 咪唑,pH 7.4)并在 20 ℃离心G 每次30秒。 最后,将树脂与 100 µl 洗脱缓冲液在热混合器中于 4 °C 下孵育 30 分钟,然后在 20 ℃ 下离心洗脱蛋白质。G 1分钟。 另加入 100 µl 洗脱缓冲液重复洗脱步骤,并将两部分洗脱液分别混合。 然后将两个洗脱液部分合并并转移至 96 孔脱盐板(货号 89807,Thermo Scientific),该板已用样品缓冲液(50 mM NaH2后4,300 mM 氯化钠,pH 7.4)。 蛋白质样品添加1后保存在-80°C⨯ 蛋白质稳定混合物(目录号 89806,Thermo Scientific)。 然后,等分10μl蛋白质并保存在-20℃作为纯化蛋白质(P)样品用于凝胶电泳。
对于第 2 轮和第 3 轮的酶以及第 2 轮预测试的截短酶,通过 Qubit Protein Assay(货号 Q33211,Thermo Scientific)测量蛋白质浓度。
凝胶电泳
每个样品的总蛋白、可溶蛋白和纯化蛋白与 1⨯加载缓冲区(4⨯上样缓冲液配方:0.2 M Tris-HCl、0.4 M DTT、277 mM SDS、6 mM 溴酚蓝、4.3 M 甘油),然后在 PCR 循环仪中于 85 °C 加热 5 分钟。 使用预制凝胶(目录号 WG1403A,Thermo Scientific)通过 SDS-PAGE 分析变性蛋白质,然后使用 InstantBlue(目录号 ISB1L-53,Kem-en-tec)进行考马斯染色。 还加载了 Spectra 多色宽范围蛋白质梯(目录号 26634,Thermo Scientific)来分析蛋白质大小。
酶法测定
要测试 MDH 活性,2 µl 或 100 µg ml−1 将第 1 轮中纯化的蛋白质加入到含有约 1.5 mM NADH(目录号 10128023001,Merck)、2.0 mM 草酰乙酸(目录号 O4126,Sigma)和 20 mM HEPES 缓冲液(pH 7.4)的反应混合物中。 在 96 孔板中一式三份进行测定。 所有组分均使用多通道移液器添加,以避免每个孔的反应时间滞后。 最终反应体积为100 µl,反应在透明96孔微孔板(目录号0020821,Sarstedt)中于室温下进行。 通过将 NADH 氧化为 NAD,一式三份测量 MDH 活性+,在 BMG Labtech SPECTROstar 纳米分光光度计中以动力学模式进行 15 分钟的 340 nm 处吸光度读数。 在无底物对照中监测 NADH 的非特异性氧化,并从其他样品中减去这些值。 使用比尔-朗伯定律将吸收值转换为 NADH 浓度 C=A/(d ⨯ e),其中消光系数e值为 6.22 mM−1厘米−1 ,以及 96 孔板中 100 μl 的路径长度 ( d) 为 0.29 厘米。 对于没有表现出任何催化活性的样品,使用十倍体积,即20μl纯化蛋白进行第二次测定。
对于第 2 轮和第 3 轮中的 MDH,20 μg ml−1 酶与阳性对照 MDH4 一起用于如上所述的测定,以定量比较催化活性,摘自第 2 轮的样品 1564 和 1546,其浓度为 0.2 μg/ml−1 由于蛋白质产量低而使用。
使用 SOD 测定试剂盒(目录号 19160,Sigma)在 96 孔格式中测量 SOD 活性,并使用多通道移液器添加所有组分,以避免每个孔的反应时间滞后。 对于第 1 轮的 SOD,将等分试样 (2 µl) 的纯化蛋白质添加到含有 98 µl 工作溶液的每个孔中。 每个样品的测定一式三份并在一个“无 XO”孔中进行。 最后将黄嘌呤氧化酶工作溶液 (10 μl) 添加到每个孔中,“无 XO”孔除外。 “无 SOD”和“空白”测定也一式三份进行。 “无 SOD”孔含有 10 µl 稀释缓冲液、80 µl 工作溶液和 10 µl 黄嘌呤氧化酶工作溶液,而“空白”孔含有 20 µl 稀释缓冲液和 80 µl 工作溶液。 将板在预设为 37°C 的读板器中孵育。 在动力学模式下测量 450 nm 处的吸光度 30 分钟。 对于没有表现出任何催化活性的蛋白质,使用十倍体积的20μl纯化蛋白质进行第二次测定。
对于第 2 轮和第 3 轮的 SOD,5 µg ml−1 如上所述的测定中使用酶以定量比较催化活性。
为了测定截短的蛋白质,85 μg ml−1 所有样品均用于酶测定。
有关溶菌酶测定的详细信息,请参阅补充方法。
数据分析
对于 MDH,随时间绘制吸光度值。 将测定终点时所有样品的吸光度值与阴性对照进行比较 t-测试分析。 如果最终吸光度值显着低于阴性对照,则样品被认为是活性的, 磷≤0.05。
对于 SOD,酶活性测量为 WST-1 甲臜形成速率的抑制百分比,并使用以下方程和 20 分钟吸光度值进行计算。 抑制率与阴性对照比较 t-测试,那些活性显着高于阴性对照的被认为是活性的磷 ≤0.05。
SOD活性(抑制率%)=((A –乙 ) − ( C – D))/( A– 乙)⨯100,其中A是“无 SOD”对照的吸光度值,乙为空白吸光度值,C是样品的吸光度值,D是“无 XO”的吸光度值。
使用 GraphPad Prism v8.0.0 for Windows、GraphPad 软件对分析数据进行分析 (www.graphpad.com)。
酶活性的半定量比较
使用 20 µg ml 进行第 3 轮酶测定的数据−1 MDH 或 5 µg/ml−1如上所述,SOD 用于酶比活性的半定量比较(图 1)。 3d)。
对于MDH,使用MDH4作为野生型阳性对照,对于SOD,使用hSOD、paSOD和E.SOD作为野生型阳性对照。
对于 MDH,将 340 nm 处的吸光度转换为 NADH 浓度,并使用测定的 0 和 90 秒时间点之间的平均浓度差作为酶活性的量度。 一些酶,包括 MDH4 对照,非常快地转化底物,使得大部分底物在第一个时间点之前被转化。 因此,我们用阴性对照的平均值替换了时间 0 时低于 275 µM 的任何值。 值是三个技术重复的平均值,并除以 MDH4 样品的平均值。
对于SOD,使用如上所述计算的抑制率(%)作为酶活性的量度。 数值取三个技术重复的平均值,并除以 hSOD、paSOD 和 E.SOD 样品的平均值。
报告摘要
2024-04-23 00:00:00
1713933085





:quality(70):focal(2072x1161:2082x1171)/cloudfront-eu-central-1.images.arcpublishing.com/liberation/7RGUHWQ6HRASVPF5V5RT4KCT6M.jpg)





